
Practical Inference Algorithms for Species Networks
ICERM - November 2024
by Hector Baños
Phylogenetics is a field of evolutionary biology dedicated to understanding the evolutionary relationships among species. Inferring these relationships is essential for diverse applications, including conservation efforts, tracking infectious disease dynamics, and improving agricultural practices.
Phylogenetics is a field of evolutionary biology dedicated to understanding the evolutionary relationships among species. Inferring these relationships is essential for diverse applications, including conservation efforts, tracking infectious disease dynamics, and improving agricultural practices.
Hybrid speciation, or hybridization, occurs when two distinct populations merge genetically to form a new population. Recent advances in genome sequencing and phylogenetic analysis have shown the significant role of hybridization in the tree of life across a wide variety of species, from orchids to leopards. As a result, there is a need for effective methods to infer hybridization, using realistic models of biological processes to better understand species' evolutionary relationships.
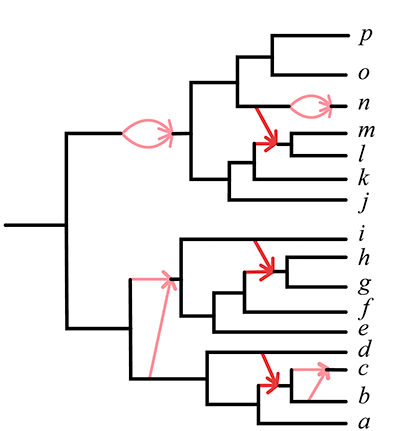
My research and the scope of my PRIMES grant focus on developing practical algorithms to uncover complex evolutionary relationships in the presence of hybridization. Traditionally, species networks are the objects used to depict evolutionary relationships when hybridization is involved (see Figure 1). A major challenge in inferring these networks is known as gene tree incongruence, where the evolutionary history of individual genes (gene trees) may not reflect that of the species. Gene tree incongruence is a central issue in modern phylogenetics and can result from naturally occurring factors such as incomplete lineage sorting, gene duplication and loss, horizontal gene transfer, and hybridization, as well as methodological errors in gene tree inference.
The theoretical foundation for my research is the Network Multispecies Coalescent (NMSC) model [1], a standard stochastic model describing the differing evolutionary histories of gene trees in the presence of both incomplete lineage sorting and hybridization. Current methods for inferring species network features under the NMSC are restricted to a rather simple class of “level-1” networks, or networks with no interlocking cycles. A goal of my research includes developing algorithms that infer statistically consistent estimators of species networks, focusing on supporting a wider range of networks and utilizing individual gene trees or genomic sequences composed of many genes as input. Our work also involves implementing these algorithms in user-friendly, reliable, and efficient software, capable of handling large datasets within practical timeframes while requiring reasonable computational resources. My research team and I have developed the R package MSCquartets for this purpose [2].
Specifically, we developed the Network inference Algorithm via NeighborNet Using Quartet distance (NANUQ), implemented in MSCquartets. NANUQ is a statistically consistent algorithm that offers a significant speed advantage over other algorithms because it eliminates the need for network search entirely, instead using combinatorial techniques to construct the network. This efficiency enables researchers to analyze large datasets whose size may be impractical for other methods. Additionally, NANUQ can detect model violations, such as when data does not originate from a level-1 network under the NMSC. Another key advantage is that NANUQ does not require prior knowledge of the number of hybridization events in a network, unlike other approaches.
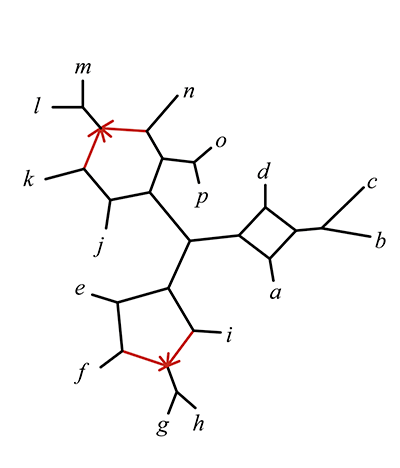
We have made significant progress in identifying more general families of networks that can be inferred from data, as well as in developing algorithms for statistically consistent estimation of species network features. This progress, along with new research directions, has accelerated notably since the start of the ICERM Semester Program. For instance, my officemate Vu Dihn (University of Delaware), whom I met at ICERM, and I began investigating the pitfalls of various inference methods under model misspecification. This research will raise evolutionary biologists’ awareness of these issues and encourage caution in their analyses.
In summary, by expanding the range of identifiable network classes, addressing the limitations of existing methods, and enhancing the reliability and scalability of species network inference techniques, our work provides biologists with the tools and insights necessary for more accurate and comprehensive investigations of evolutionary relationships.
Hector Baños is a long-term participant in ICERM’s Theory, Methods, and Applications of Quantitative Phylogenomics Semester Program and a U.S. National Science Foundation Partnerships for Research Innovation in the Mathematical Sciences (PRIMES) awardee. The PRIMES program seeks to support relationships between NSF Division of Mathematical Sciences-supported institutes such as ICERM and faculty at minority-serving institutions.
References
[1] Chen Meng and Laura Salter Kubatko. Detecting hybrid speciation in the presence of incomplete lineage sorting using gene tree incongruence: A model, Theoretical Population Biology, 75(1): 35-45, 2009. URL: https://doi.org/10.1016/j.tpb.2008.10.004.
[2] John A Rhodes, Hector Baños, Jonathan D Mitchell, and Elizabeth S Allman. MSCquartets 1.0: quartet methods for species trees and networks under the multispecies coalescent model in R, Bioinformatics, 37(12):1 766-1768, 2021. URL: https://doi.org/10.1093/bioinformatics/btaa868.
[3] C Solís-Lemus and Cécile Ané. Inferring Phylogenetic Networks with Maximum Pseudolikelihood under Incomplete Lineage Sorting, PLOS Genetics, 12(3), 2016. URL: https://doi.org/10.1371/journal.pgen.1005896.