
Our Changing World: Algebraic Statistics for Evolutionary Biology and Ecology
IMSI - June 2024
Understanding the evolutionary history of a collection of species, through fields such as phylogenomics and comparative phylogenetics, is crucial as we consider the future effects of climate change. For example, these fields provide insights into the roles different species play in their environments, which is essential for predicting how ecosystems might change under different climate scenarios and the cascading effects on biodiversity and human societies.
Algebraic statistics provides algebraic and geometric tools to study the models commonly used in evolutionary biology. The Institute for Mathematical and Statistical Innovation (IMSI), hosted a workshop, “Algebraic Statistics for Ecological and Biological Systems,” as part of a Long Program on “Algebraic Statistics and Our Changing World,” which highlighted these connections. John Rhodes (University of Alaska, Fairbanks), began the workshop with a presentation titled “Inferring the tree-like parts of a species network under the coalescent.” Rhodes’s talk highlighted his group’s recent work investigating the inference of evolutionary histories using genomic data under the network multispecies coalescent model (NMSC). An NMSC of a population is useful when the population’s genetic history is not fully known. It allows for the creation of a model of the population’s genetics that can help understand the inheritance of genes and how species differentiated over time. The creation of these models and the predictions that follow are very challenging. Algebraic statistics provides mathematical tools that ease use of the NMSC.
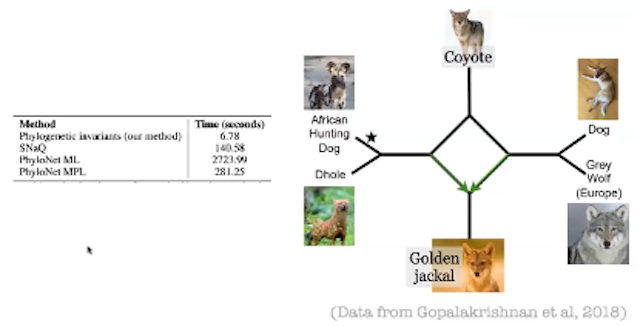
In the same workshop, Claudia Solis Lemus (University of Wisconsin – Madison) presented a talk entitled, “Ultrafast learning of hybridization networks using phylogenetic invariants.” Solis Lemus illustrated the accuracy and speed of her group’s new algebra-based method on a collection of simulated phylogenetic trees, and then used this method to estimate the phylogenetic network for the genus Canis (dogs, coyotes, wolves, etc). Solis Lemus demonstrated that her group’s new methodology is 20 to 400 times faster at reconstructing phylogenetic networks as compared to other accepted methods (See the thumbnail image and its caption below).
The best way to test the predictions in NMSC models and choosing which model works best was a main research theme of the semester program with a semester working group focusing on how to apply a recent model selection technique in algebraic statistics from the paper, “Testing Many Constraints in Possibly Irregular Models Using Incomplete U-Statistics” by Nils Sturma, Mathias Drton, and Dennis Leung to phylogenetic data. The working group included a presentation by Nils Sturma (Technical University of Munich) on the paper and brought together junior and senior researchers working in mathematical phylogenetics.
On the comparative phylogenetics side, Chris Muir (University of Wisconsin – Madison) discussed, “How empiricists use phylogenetic comparative methods to study trait evolution.” He explained how convergence among lineages toward similar traits in similar circumstances is powerful evidence for biological adaptation; and issues such as identifiability, parameter interpretation, model adequacy, models of complex traits, and model comparison could use more theoretical treatment. Algebraic geometry has a role to play in the analysis of these models.
One of the more important outcomes of this Long Program was that it brought together a diverse community of scholars who added a new level of mathematical insight and interdisciplinarity to this relatively new field of research.
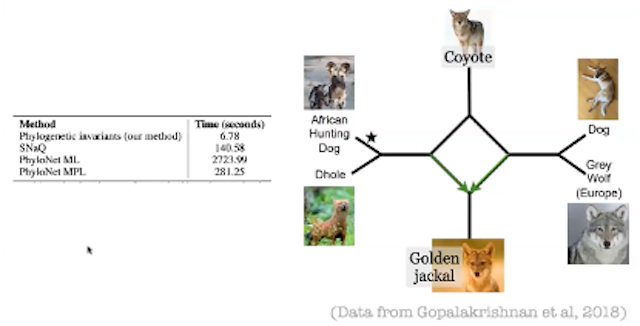
Image caption: Phylogenetic network of the genus Canis, with a comparison of the estimation times of various methodologies. “Phylogenetic invariants” is the methodology of Solis Lemus and her collaborators.