
Algebraic Tools for Phylogenetic Networks
ICERM - April 2025
by Seth Sullivant (North Carolina State University)
Based on joint work with Marta Casanellas, Universitat Politècnica de Catalunya; Aviva Englander, University of Wisconsin, Madison; Jesús Fernández-Sánchez, Universitat Politècnica de Catalunya; Martin Frohn, Maastricht University; Elizabeth Gross, University of Hawai‘i at M¯anoa; Benjamin Hollering, TU Munich; Niels Holtgrefe, TU Delft; Leo van Iersel, TU Delft; and Mark Jones, Centrum Wiskunde & Informatica
I was a long term visitor at ICERM during the Fall 2024 program “Theory, Methods, and Applications of Quantitative Phylogenomics". It was an excellent sabbatical visit for me, and in the 6 weeks I was at ICERM I started three new projects with a range of collaborators [2, 3, 7]. All three of these projects are unified by the study of phylogenetic network models and using tools from algebraic geometry to address their study.
Phylogenetics is the field of mathematical biology concerned with recovering and describing evolutionary relationships between collections of taxa. Taxon is short for taxonomic unit and could refer to a species, a gene, a language or other basic object that undergoes an evolutionary process. The branching trajectory of evolution is often represented via a phylogenetic tree: vertices on the tree represent different taxa and taxa that are close to each other in the tree are evolutionarily close to each other. A major theme at the ICERM Fall 2024 program concerned the effort to move beyond trees. Many groups of species undergo hybridization (the formation of a new species from the shared genetic material of two other species), and so modeling evolutionary processes using an underlying network structure (and not just a tree), is seen as increasingly important. However, the resulting phylogenetic network models are significantly more complicated than models for phylogenetic trees.
One of the most basic questions to address about a new family of models is whether or not the parameters in these models are identifiable from the model observed quantities. There are many types of model structure used in phylogenetic network models as well as many types of observed quantities. We employed algebraic tools to study identifiability in the case of a model called the network-based Markov model or the displayed trees model where the observed data consist of the DNA sequences that are produced. This is in contrast to other results that focus on the Network Multispecies Coalescent and use other types of data like observed gene trees or concordance factors [1]. Past results on the identifiability of phylogenetic networks have focused on identifiability for level 1-networks [5], or to identify the tree of blobs of a more complex network [1].
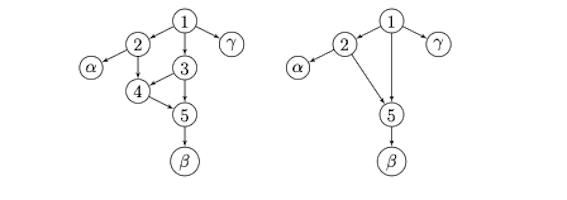
A reticulation in a network is a node that represents a hybridization event: this is where 2 or more edges enter a vertex. A network has level k if each 1-connected component has at most k reticulation vertices. Figure 1 shows examples of level 1 and level 2 networks. In both networks in the figure, node 5 is a reticulation, and 4 is also a reticulation in the network on the right. Level-1 networks are hence very simple in that they can only have cycles that are not connected to each other. Generally, one expects more complex hybridization than can be represented with a level-1 network and so data analysis that assumes networks of higher level need to be developed. To get to new algorithms to construct higher level networks from data, we first need identifiability results about higher level networks.
Aviva Englander, Martin Frohn, Elizabeth Gross, Niels Holtgrefe, Leo van Iersel, Mark Jones, and I set out to try to prove identifiability results beyond level-1 networks. The collaboration presents a combination of researchers with perspectives coming from combinatorics and algebraic geometry to address problems in phylogenetics. This is a collaboration that started at ICERM and benefited greatly from the group working environment present at the institute. Our main result is the first identifiability result on level-2 networks [3]:
Theorem 1. The network parameter of a network-based Markov model under the Jukes-Cantor constraints is generically identifiable when the network parameter is an n-leaf binary, triangle free, strongly tree-child, level-2 semi-directed phylogenetic network.
You might be wondering: "Why all the adjectives?" Some of these conditions are related to known nonidentifiability issues. For example, semi-directed appears because the root of the network is not identifiable. Some of these conditions are related to limitations in our techniques, and represent aspirations for proving stronger results. Specifically triangle-tree represents a condition that is often present in identifiability results for networks, but practitioners would love to eliminate it because hybridizations often occur between closely related species, which create triangles in networks. And some of these conditions represent surprising new situations where we have found global non-identifiable results. In particular, strongly tree-child is in part added because it rules out stacked reticulations, which our results show are never identifiable in network-based Markov models [7]. A stacked reticulation is a pair of reticulations where one reticulation is a child of another. So the network on the right in Figure 1 the pair of vertices 4 and 5 form a stacked reticulation. In fact, the two networks in this figure always produce the same family of probability distributions under the network-based Markov model for any nice Markov model.
Finally, a new project was started with Marta Casanellas, Jesús Fernández-Sánchez, Elizabeth Gross, and Ben Hollering which proposes to push our algebraic geometric understanding of phylogenetic network models. Previous results [4, 5] and our recent result [3] focus on the group based phylogenetic models (which include the Jukes-Cantor, and Kimura models). The group based structure allows for great simplifications on both the computational and theoretical side of the study. In [2], we are pushing results for identifiability of level-1 networks and to recover blob trees to the larger class of equivariant phylogenetic models, including new identifiability results for the General Markov Model.
All three of these projects [2, 3, 7] embrace the perspective at ICERM of computational and experimental mathematics. One of my goals in attending the Fall 2024 ICERM program had been to find applications of a technique that Ben Hollering and I had developed using algebraic matroids to prove identifiability results [6]. This method is fundamentally a strategy of doing large scale randomized computations to find an algebraic certificate of identifiability. The beginning stages of these projects involved lots of symbolic and numerical computations, and the end results in both [2, 7] have proofs that depend in part on computations.
References
[1] Elizabeth S. Allman, Hector Baños, Jonathan D. Mitchell, and John A. Rhodes, The tree of blobs of a species network: identifiability under the coalescent, J. Math. Biol. 86 (2023), no. 1, Paper No. 10, 26. MR 4519611
[2] Marta Casanellas, Jesús Fernández-Sánchez, Elizabeth Gross, Benjamin Hollering, and Seth Sullivant, Phylogenetic networks evolving under g-equivariant models, In preparation, 2025.
[3] Aviva Englander, Martin Frohn, Elizabeth Gross, Niels Holtgrefe, Mark Jones, Leo van Iersel, and Seth Sullivant, Identifiability of phylogenetic level-2 networks under the jukes-cantor model, In preparation, 2025.
[4] Elizabeth Gross and Colby Long, Distinguishing phylogenetic networks, SIAM J. Appl. Algebra Geom. 2 (2018), no. 1, 72–93. MR 3763914
[5] Elizabeth Gross, Leo van Iersel, Remie Janssen, Mark Jones, Colby Long, and Yukihiro Murakami, Distinguishing level-1 phylogenetic networks on the basis of data generated by Markov processes, J. Math. Biol. 83 (2021), no. 3, Paper No. 32, 24. MR 4311049
[6] Benjamin Hollering and Seth Sullivant, Identifiability in phylogenetics using algebraic matroids, J. Symbolic Comput. 104 (2021), 142–158. MR 4180121
[7] Seth Sullivant, Phylogenetic network models as directed acyclic graphs, In preparation, 2025.